武汉病毒研究所王延轶近期在人巨细胞病毒的免疫逃逸机制上再取新进展

(图片来源:摄图网)
作者|病毒学 来源|生命科学前沿(ID:advancedlifesci)
人巨细胞病毒(HCMV)含有一个大的双链DNA,编码的蛋白超过200多种,是疱疹病毒家族的一员。HCMV是一种条件性病原体,在免疫力强的个体中几乎不会引起严重疾病,但新生儿感染HCMV可导致中枢神经系统不可逆性损伤,免疫低下的器官移植者或AIDS患者感染后可引起严重综合征。
先天性免疫反应是宿主抵抗病毒感染的第一道防线。宿主细胞的模式识别受体(PRR)识别病原体的病原体相关分子模式(PAMP),引发细胞信号级联反应,调节机体产生I型干扰素和促炎因子,进而大范围的触发机体抗病毒反应,包括抑制病毒复制,清除病毒感染的细胞和激活适应性免疫应答。
病毒核酸(DNA或RNA)是一种强力的PAMP,能被PRR识别。环鸟苷酸-腺苷酸(cGAMP)合成酶(cGAS)是一种普遍存在细胞中的DNA感受蛋白。一旦监测到胞质中存在外源DNA,cGAS会利用鸟苷三磷酸(ATP)和腺苷三磷酸(GTP)合成细胞内第二信使cGAMP。
随后cGAMP结合并激活内质网相关的衔接蛋白 MITA(也称为 STING)促进其发生二聚化,活化状态的 MITA 经内质网、高尔基体转移到细胞核周围点状聚集体或微粒体,转移过程中募集其他蛋白比如激酶TBK1和干扰素调节因子3(IRF3,一种转录因子),随之被TBK1磷酸化的IRF3从细胞质进入细胞核诱导干扰素和一些细胞因子的基因表达。
4月1日武汉病毒所所长王延轶与副研究员付玉志以共同通讯作者身份在《Journal of Virology》发表了题为“Human Cytomegalovirus Protein UL94 Targets MITA to Evade Antiviral Immune Response”的文章,揭示了人巨细胞病毒的免疫逃逸机制。
在此,我们对该论文的主要结果进行了概要总结:
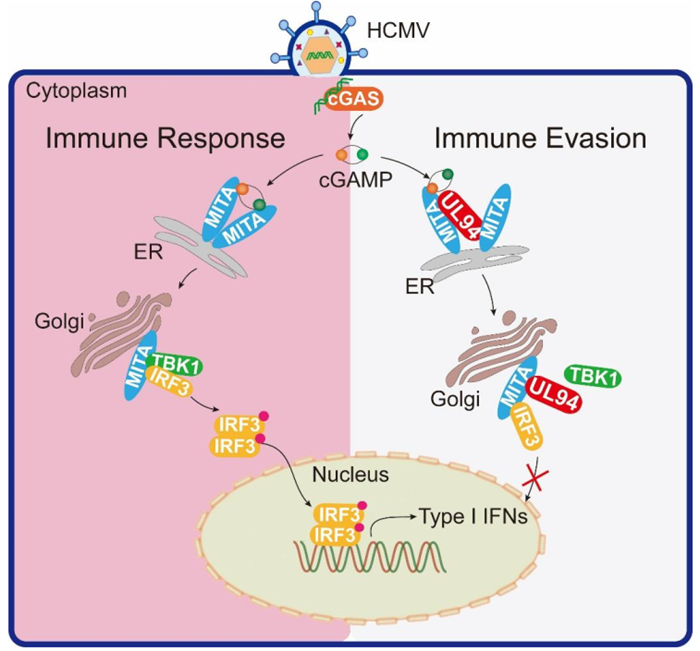
1. 通过在哺乳动物细胞中单独表达不同的HCMV蛋白,利用和检测cGAS-MITA信号下游的报告基因,鉴定到HCMV被膜蛋白UL94能抑制cGAS-MITA介导的相关基因的表达;
2. 使用人原代包皮成纤维细胞(HFF),研究人员再次验证了UL94可抑制一些抗病毒基因的表达,而且观察到 UL94能抑制 TBK1和 IRF3的磷酸化;
3. 与上述结果一致,研究人员在感染HCMV的HFF细胞中敲低UL94后,HCMV诱导的相关基因的表达及TBK1和 IRF3的磷酸化水平都增加;
4. UL94是病毒感染晚期表达的一种结构蛋白,TCID50实验(反映病毒滴度)发现UL94能增加感染晚期HCMV在野生型HFF细胞中的复制,而在MITA敲除的HFF细胞中其复制受阻,揭示了UL94通过破坏MITA信号来增强HCMV复制能力;
5. 随后证实UL94仅与信号通路中的MITA组分(不与TRAPβ, iRhom2 和SNX8结合)直接作用来抑制机体的抗病毒先天免疫反应;
6. 进一步分析发现UL94破坏了MITA自身的二聚化,进而影响其向核周微粒体的转移;
7. 同时发现UL94削弱了MITA与TBK1的结合,导致下游信号转导发生抑制。总之,这些发现揭示了UL94在HCMV的免疫逃逸中起重要作用,拓宽了我们对HCMV感染晚期发生免疫逃逸的认识。
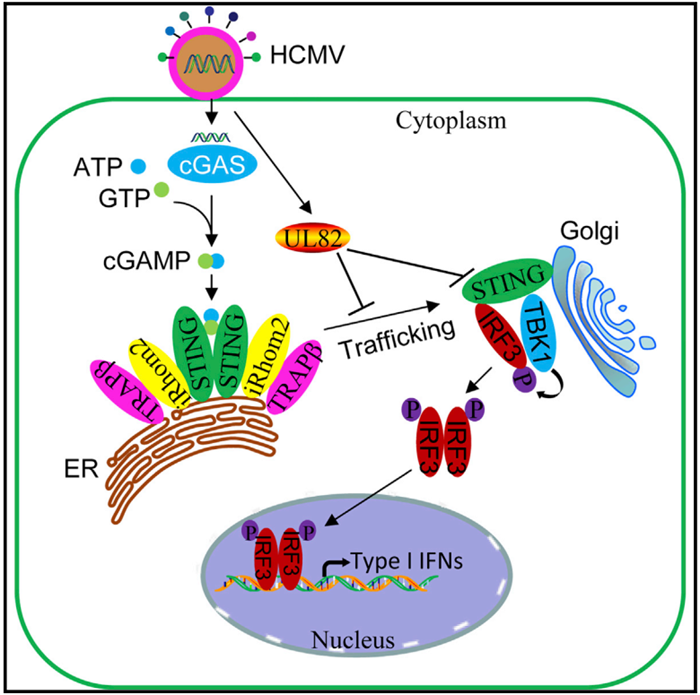
早在2017年2月份,武汉大学副校长舒红兵作为唯一通讯作者,王延轶共同参与的研究也揭示了被膜蛋蛋白在HCMV免疫逃逸中的重要作用。
该项题为“Human Cytomegalovirus Tegument Protein UL82 Inhibits STING-Mediated Signaling to Evade Antiviral Immunity”的文章发表于《Cell Host & Microbe》。
研究人员筛选出UL82 可显著抑制宿主感染HCMV后的抗病毒免疫反应;UL82 缺陷型HCMV 诱导的宿主抗病毒基因表达水平高于野生型 HCMV,即野生型 HCMV具有更强的复制能力。
进一步探索发现UL82能够与STING 和另一个组分iRhom2相互作用,从而抑制STING-TRAPβ/iRhom2-TRAPβ的联系,破坏STING-iRhom2-TRAPβ 易位复合物形成,导致 STING 无法从内质网向核周围转移, 同时UL82 也削弱了MITA 对TBK1和IRF3募集。
这些发现共同揭示了HCMV发生免疫逃避的一个重要机制,为研发抵抗HCMV的药物和疫苗提供了重要理论依据。
编者按:本文转载自微信公众号:生命科学前沿(ID:advancedlifesci)


本文作者信息
生命科学前沿(科学自媒体)
邀请演讲广告、内容合作请点这里:寻求合作
咨询·服务